Efficacy in R/R
NPM1-m AML with
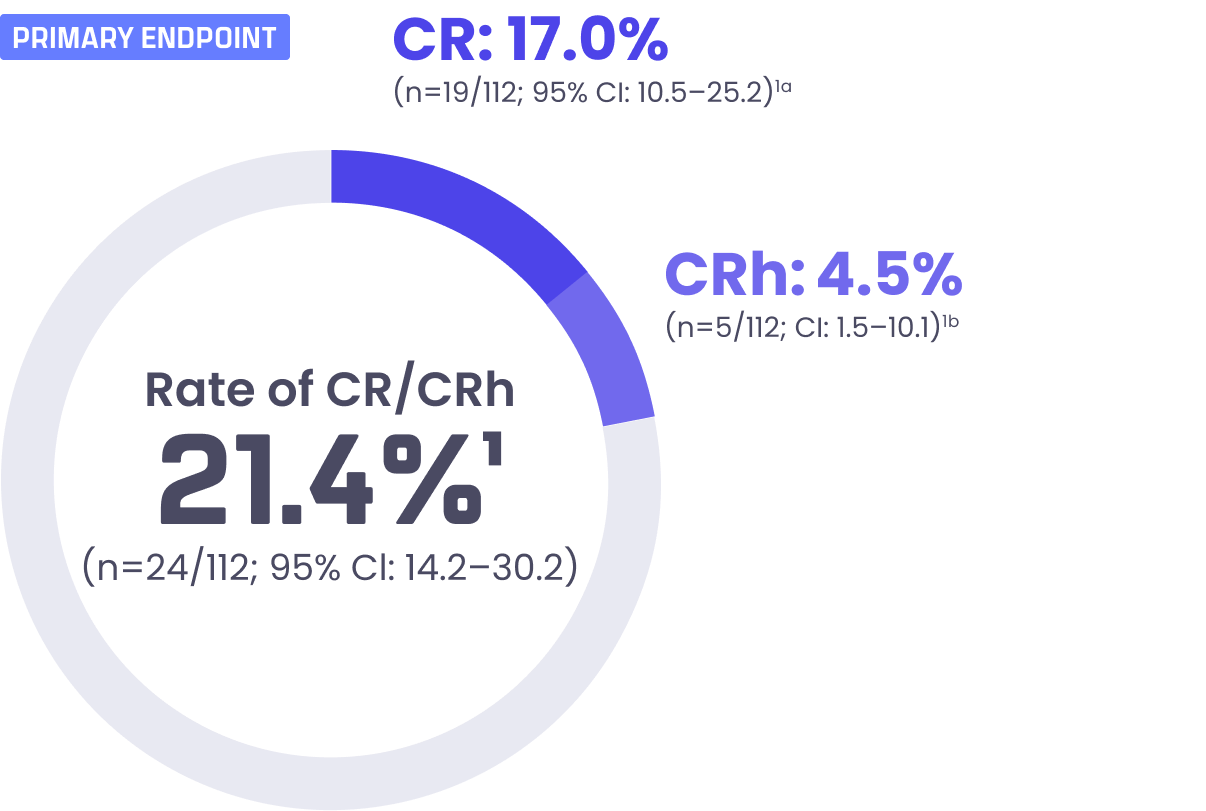
KOMZIFTI
KOMET-001 is the largest pivotal monotherapy study of an approved menin inhibitor in adults with
R/R NPM1-m AML1,2
KOMET-001 was a Ph1b/2, open-label, single-arm study1,2
One-third of trial participants received KOMZIFTI as early as 2L3
KOMZIFTI delivered meaningful remission
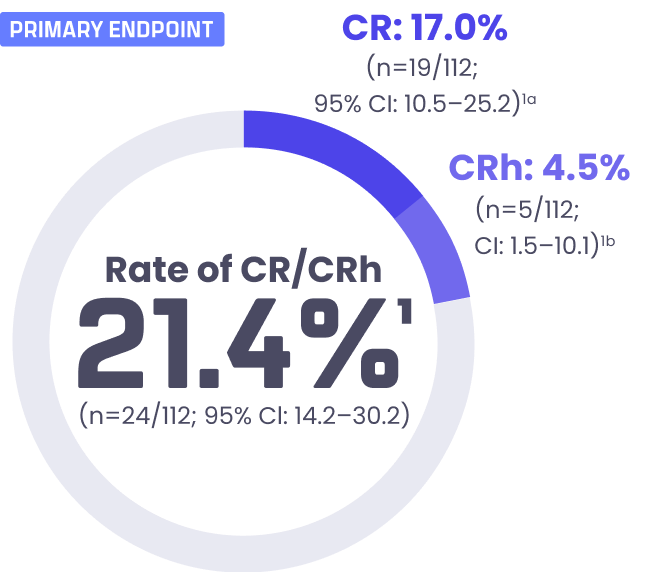
- Median duration of CR/CRhc: 5.0 months (95% CI: 1.9-8.1)1
- Median follow-up: 4.2 months (range: 0.1-41.2 months)1
achieved MRD negativity among assessed CR/CRh responders
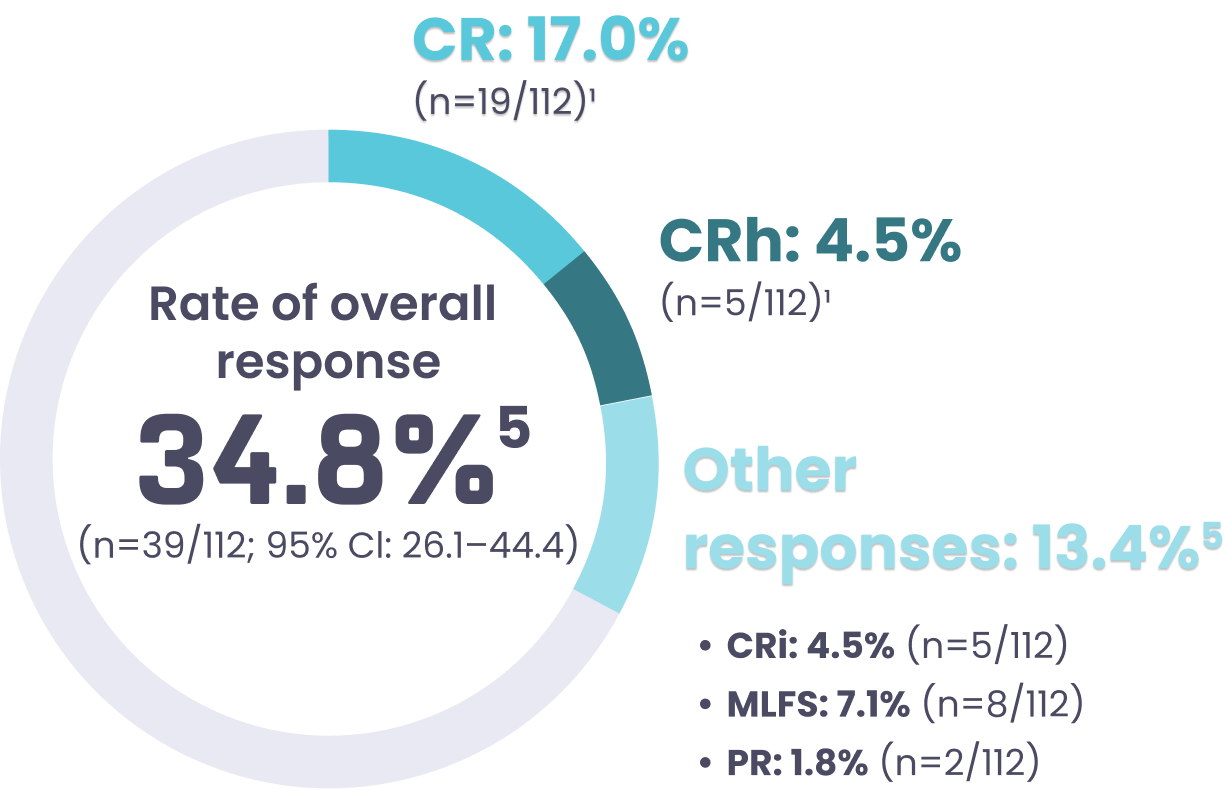
Overall responsee rate
with KOMZIFTI

- Median duration of response: 4.6 months (95% CI: 3.7–6.0; range: 0.0+–20.5+)5
(n=4/39) of responders underwent HSCT following KOMZIFTI treatment6f
- aCR is defined as bone marrow blasts <5%, ANC >1.0 x 109/L, and platelet count >100 x 109/L.1
- bCRh is defined as bone marrow blasts <5%, ANC >0.5 x 109/L, and platelet count >50 x 109/L.1
- cDuration of CR/CRh is defined as the time from first CR/CRh to the first documented relapse or death, whichever occurs first.1
- dAssessment among responders with samples evaluable for MRD by central laboratory testing. MRD negativity is defined as achieving at least one post-baseline MRD negative result.2,3
- eIn KOMET-001, response was assessed for Ph1b using 2017 ELN AML criteria (which included CR, CRh, CRi, CRp, MLFS, and PR) and for Ph2 using 2022 ELN AML criteria (which included CR, CRh, CRi, MLFS, and PR).5
- f3.6% of trial participants underwent HSCT following KOMZIFTI.1
Consider KOMZIFTI as early as 2L1
33% (n=37/112) of patients had 1 prior line of therapy in the KOMET-001 study3
CR/CRh rates across select prespecified patient subgroups1,2,4
Small patient numbers and lack of multiplicity adjustments can be limitations of these analyses. No efficacy conclusions can be drawn from these data.

Small patient numbers and lack of multiplicity adjustments can be limitations of these analyses. No efficacy conclusions can be drawn from these data.

All subgroups were prespecified with the exception of DNMT3A co-mutational status, which was only applied to the Phase 2 mITT analysis set; results should be interpreted with caution.2,4
For patients without confirmed disease progression or unacceptable toxicity:
To allow time for a clinical response, continue KOMZIFTI for a minimum of 6 months1

- Among CR/CRh responders, the median time to first responsea ranged from 1 to 15 months1
KOMZIFTI is a menin inhibitor that promotes cell differentiation1
aResponse assessments were conducted at the end of each 28-day treatment cycle, and response metrics were defined according to the 2022 European LeukemiaNet Recommendations for AML.2
Explore the safety profile of KOMZIFTI
Transfusion independence was possible with KOMZIFTI1
Independence achieved1
Of the 66 patients who were dependent on RBC and/or platelet transfusions at baseline, 14 (21%) became independent of RBC and platelet transfusions during any 56-day post-baseline period.
Independence maintained1
Of the 46 patients who were independent of both RBC and platelet transfusions at baseline, 12 (26%) remained transfusion independent during any 56-day post-baseline period.
The following is a post-hoc analysis. Findings are exploratory in nature and not prespecified in the original study protocol; no conclusions should be drawn from these data.

aTransfusion conversion rate is defined as the number of patients who were TD at baseline but become TI post-baseline (ie, n) divided by the total number of patients who were TD at baseline (ie, m). Patients were TI post-baseline if they had a transfusion-free period of at least 56 consecutive days during the post-baseline period.7
KOMZIFTI is the first and only oral menin inhibitor approved for once daily use1
Learn About Dosing for KOMZIFTIKOMET-001 is the largest pivotal monotherapy study of an approved menin inhibitor in adults with R/R NPM1-m AML1
KOMET-001: Ph1b/2 open-label, single-arm study

Select inclusion criteria
- Presence of a susceptible NPM1 mutation
- Creatinine clearance ≥30 mL/min
- Total bilirubin ≤1.5 x ULN
- Aminotransferases ≤2 x ULN
- QTcF ≤480 msec
- ECOG Performance Status score 0–2
Select secondary endpoints
- Duration of CR/CRh
- ORR
- Time to response,
duration of response
- MRD negativity
- OS
- Transfusion independence
- Safety and tolerability
Select baseline characteristics and prior treatment history
Select baseline characteristics
63%
Population age ≥65 years1
- Median age: 69 years (range: 22–86)1
45%
FLT3 Co-mutations4
83%
ECOG PS: 0 or 13a
Select prior
treatment history
60%
Prior Venetoclax Exposure3a
33%
1 Prior Therapy3a
- Median prior therapies:
2 (range: 1–7)1
23%
Prior HSCT1


aResults from Wang, 2025 reflect an earlier data cut; outcomes are consistent with those reported in Prescribing Information.
bDNMT3A only applied to Phase 2 mITT set.4
Primary refractory: persistent leukemia following induction.8
Refractory relapse: unresponsive to most recent salvage treatment.8
Untreated relapse: initial response to most recent treatment with subsequent relapse.8
aResults from Wang, 2025 reflect an earlier data cut; outcomes are consistent with those reported in Prescribing Information.
AML, acute myeloid leukemia; ANC, absolute neutrophil count; CI, confidence interval; CR, complete remission; CRh, complete remission with partial hematologic recovery; CRi, complete remission with incomplete hematologic recovery; CRp, complete remission with incomplete platelet recovery; DNMT3A, DNA methyltransferase 3A; DNMT3Am, mutated DNA methyltransferase 3A; ECOG PS, Eastern Cooperative Oncology Group Performance Status; ELN, European LeukemiaNet; FLT3, FMS-like tyrosine kinase 3; HSCT, hematopoietic stem cell transplantation; IDH1-m, mutated isocitrate dehydrogenase 1; IDH2, isocitrate dehydrogenase 2; ITD, internal tandem duplication; MLFS, morphologic leukemia-free state; MRD, measurable residual disease; NPM1-m, mutated nucleophosmin 1; ORR, overall response rate; OS, overall survival; Ph, phase; PR, partial response; QTcF, Fridericia–corrected QT interval; RBC, red blood cell; R/R, relapsed or refractory; TKD, tyrosine kinase domain; ULN, upper limit of normal; VEN, venetoclax; +, censor.
References: 1. KOMZIFTI [Prescribing Information]. San Diego, CA; Kura Oncology, Inc. 2. Wang ES et al. J Clin Oncol. 2025 Nov;43(31):3381-3390. 3. Wang ES et al. J Clin Oncol. 2025 Nov;43(31)(suppl):3381-3390. 4. Data on File. Zifto 007. San Diego, CA; Kura Oncology, Inc. 5. Data on File. Zifto 001. San Diego, CA; Kura Oncology, Inc. 6. Data on File. Zifto 006. San Diego, CA; Kura Oncology, Inc. 7. Data on File. Zifto 003. San Diego, CA; Kura Oncology, Inc. 8. Döhner H et al. Blood. 2022;140(12):1345-1377.
Discover the safety profile of KOMZIFTI and
guidelines for certain adverse reactions
Review Safety Information for KOMZIFTI
WARNING: DIFFERENTIATION SYNDROME
Differentiation syndrome, which can be fatal, has occurred with KOMZIFTI. Signs and symptoms may include fever, joint pain, hypotension, hypoxia, dyspnea, rapid weight gain or peripheral edema, pleural or pericardial effusions, pulmonary infiltrates, acute kidney injury, and rashes. If differentiation syndrome is suspected, interrupt KOMZIFTI, and initiate oral or intravenous corticosteroids with hemodynamic and laboratory monitoring until symptom resolution; resume KOMZIFTI upon symptom improvement.
WARNINGS AND Precautions
Differentiation Syndrome
KOMZIFTI can cause fatal or life-threatening differentiation syndrome (DS). DS is associated with rapid proliferation and differentiation of myeloid cells. Symptoms of DS, including those seen in patients treated with KOMZIFTI, may include fever, hypoxia, joint pain, hypotension, dyspnea, rapid weight gain or peripheral edema, pleural or pericardial effusions, acute kidney injury, and rashes.
In the clinical trial, DS occurred in 29 (26%) of 112 patients with R/R AML with an NPM1 mutation who were treated with KOMZIFTI at the recommended dosage. DS was Grade 3 in 13% and fatal in two patients. In broader evaluation of all patients with any genetic form of AML treated with
KOMZIFTI monotherapy in clinical trials, DS occurred in 25% of patients. Four fatal cases of DS occurred out of 39 patients with KMT2A-rearranged AML treated with KOMZIFTI. KOMZIFTI is not approved for use in patients with KMT2A-rearranged AML.
In the 112 patients with an NPM1 mutation, DS was observed with and
without concomitant hyperleukocytosis, in as early as 3 days and up to
46 days after KOMZIFTI initiation. The median time to onset was 15 days.
Two patients experienced more than one DS event. Treatment was interrupted and resumed in 15 (13%) patients, while it was permanently discontinued in 2 (2%) patients.
Prior to starting treatment with KOMZIFTI, reduce the WBC counts to less than 25 x 109/L. If DS is suspected, interrupt KOMZIFTI, initiate oral or intravenous corticosteroids (e.g., dexamethasone 10 mg every 12 hours) for a minimum of 3 days with hemodynamic and laboratory monitoring. Resume treatment with KOMZIFTI at the same dose level when signs and symptoms improve and are Grade 2 or lower. Taper corticosteroids over a minimum of 3 days after adequate control or resolution of symptoms. Symptoms of DS may recur with premature discontinuation of corticosteroid treatment.
QTc Interval Prolongation
KOMZIFTI can cause QTc interval prolongation. In the clinical trial, QTc interval prolongation was reported as an adverse reaction in 12% of 112 patients treated with KOMZIFTI at the recommended dosage for R/R AML with an NPM1 mutation. QTc interval prolongation was Grade 3 in 8% of patients. The heart-rate corrected QT interval (using Fridericia’s method) (QTcF) was greater than 500 msec in 9% of patients, and the increase from baseline QTcF was greater than 60 msec in 12% of patients. KOMZIFTI dose reduction was required for 1% of patients due to QTc interval prolongation. QTc prolongation occurred in 14% of the 42 patients less than 65 years of age and in 10% of the 70 patients 65 years of age or older.
Correct electrolyte abnormalities, including hypokalemia and hypomagnesemia, prior to treatment with KOMZIFTI. Perform an ECG prior
to initiation of treatment with KOMZIFTI, and do not initiate KOMZIFTI in patients with QTcF > 480 msec. Perform an ECG at least once weekly for the first four weeks on treatment, and at least monthly thereafter. Interrupt KOMZIFTI if the QTc interval is > 500 ms or the change from baseline is > 60 ms (Grade 3). In patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities, or those who are taking
medications known to prolong the QTc interval, more frequent ECG monitoring may be necessary. Concomitant use of KOMZIFTI with drugs known to prolong the QTc interval may increase the risk of QTc interval prolongation, result in a greater increase in the QTc interval and adverse reactions associated with QTc interval prolongation, including Torsades de pointes, other serious arrhythmias, and sudden death.
Embryo-Fetal Toxicity
Based on findings in animals and its mechanism of action, KOMZIFTI can cause embryo-fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with KOMZIFTI and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with KOMZIFTI and for 3 months after the last dose.
ADVERSE REACTIONS
Fatal adverse reactions occurred in 4 (4%) patients who received KOMZIFTI, including 2 with differentiation syndrome, 1 with infection, and 1 with sudden death. Serious adverse reactions were reported in 79% of patients who received KOMZIFTI. Serious adverse reactions occurring in ≥ 5% of patients included infection without an identified pathogen (29%), febrile
neutropenia (18%), bacterial infection (16%), differentiation syndrome (16%), and dyspnea (6%).
Dosage interruption of KOMZIFTI due to an adverse reaction occurred in
54% of patients. Adverse reactions that required dose interruption in ≥ 2%
of patients included infection without an identified pathogen (15%), differentiation syndrome (13%), febrile neutropenia (5%), pyrexia (4%), electrocardiogram QT prolonged (4%), leukocytosis (4%), bacterial
infection (3%), cardiac failure (2%), cholecystitis (2%), diarrhea (2%),
pruritus (2%), and thrombosis (2%). Dose reduction of KOMZIFTI due to an adverse reaction occurred in 4% of patients. Permanent discontinuation of KOMZIFTI due to an adverse reaction occurred in 21% of patients. Adverse reactions that required permanent discontinuation of KOMZIFTI in ≥ 2% of patients were infection without an identified pathogen (8%), bacterial infection (4%), cardiac arrest (2%), and differentiation syndrome (2%).
Most common (≥ 20%) adverse reactions, including laboratory abnormalities, were aspartate aminotransferase increased (53%), infection without an identified pathogen (52%), potassium decreased (52%), albumin decreased (51%), alanine aminotransferase increased (50%), sodium decreased (49%), creatinine increased (45%), alkaline phosphatase increased (41%), hemorrhage (38%), diarrhea (36%), nausea (35%), fatigue (34%), edema (30%), bacterial infection (28%), musculoskeletal pain (28%), bilirubin increased (27%), potassium increased (26%), differentiation syndrome (26%), pruritus (23%), febrile neutropenia (22%), and transaminases increased (21%).
DRUG INTERACTIONS
Drug interactions may occur when KOMZIFTI is concomitantly used with:
- Strong or Moderate CYP3A4 Inhibitors: Monitor patients more frequently
for KOMZIFTI-associated adverse reactions. - Strong or Moderate CYP3A4 Inducers: Avoid concomitant use of KOMZIFTI.
- Gastric Acid Reducing Agents: Avoid concomitant use of KOMZIFTI with proton pump inhibitors (PPIs), H2 receptor antagonists (H2RAs), or locally acting antacids. If concomitant use with H2RAs or locally acting antacids cannot be avoided, modify KOMZIFTI administration time.
- Take KOMZIFTI 2 hours before or 10 hours after administration of an H2
receptor antagonist. - Take KOMZIFTI 2 hours before or 2 hours after administration of a locally acting antacid.
- Take KOMZIFTI 2 hours before or 10 hours after administration of an H2
- Drugs that Prolong the QT Interval: Avoid concomitant use of KOMZIFTI. If concomitant use cannot be avoided, obtain ECGs when initiating, during concomitant use, and as clinically indicated. Interrupt KOMZIFTI if the QTc interval is > 500 ms or the change from baseline is > 60 ms.
USE IN SPECIFIC POPULATIONS
Pregnancy: Based on findings in animals and its mechanism of action, KOMZIFTI can cause embryo-fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Verify pregnancy status in females of reproductive potential prior to starting KOMZIFTI.
Lactation: Because of the potential for adverse reactions in the breastfed child, advise women not to breastfeed during treatment with KOMZIFTI and for 2 weeks after the last dose.
Infertility: Based on findings in animals, KOMZIFTI may impair fertility in females and males of reproductive potential.
INDICATION
KOMZIFTI is indicated for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (R/R AML) with a susceptible nucleophosmin 1 (NPM1) mutation who have no satisfactory alternative treatment options.
Please see full Prescribing Information, including Boxed WARNING, for additional information.
